Menu items
File menu
Load
Load data from the specified server or the local file system. See the "Starting AlignmentViewer " section.
Properties
Configure user property parameters. See the "Configuring AlignmentViewer " section.
Search menu
Mark genes
Mark a specified set of genes.
An input form is opened for users to enter a list of gene names to be marked. Gene names can be entered individually or be read from a file. By pressing the Search button, gene names are registered. Locations of the registered genes are indicated on each axis of the dotplot display, and a new table window is opened containing the information of the these genes.
Additional calls of this function will add genes to the existing list. To remove genes from the list, enter the gene names to be removed and press Clear. To clear the entire list, press All Clear.

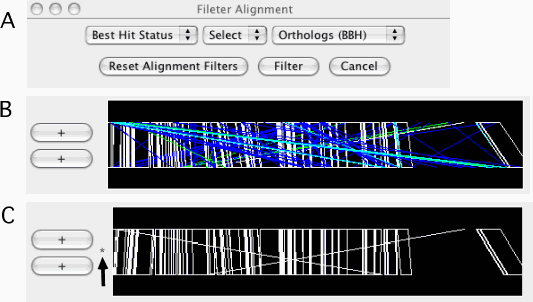
Filter Alignments
Filtering out the alignments that do not satisfy the specified conditions. Alignments are filtered from both the alignment and dotplot displays. A filter can be applied to one of the following fields:
Identity: percentage identity; numeric.
Score: similarity score; numeric.
Best Hit Status: one of the following: 3, orthologs (bidirectional best hits, BBH); 2, duplicated only in the sequence-2; 1, duplicated only in the sequence-1; 0, paralogs.
Once filtered, a '*' mark is displayed on the left side of the alignment track (see Figure 11C). Additional calls will add filters to the existing one. For example, users can specify conditions such as "Best Hit Status is BBH and Identity ≥ 90%" by two successive calls of this function. To reset the filters, press the Reset Alignment Filters button.
Filter Genes/Segments
Filtering out the genes or feature segments that do not satisfy the specified conditions. A filter can be applied to any field of any kind of segment that is currently loaded.
Kinds of conditions to be applied are different depending on the types of the fields (numeric and string). The following conditions are defined:
- Numeric field
Available conditions are: Between (a ≤ x ≤ b), Except Between(x < a or b < x), Less Than (x < a), Less Equal (x ≤ a), Equal (x = a), Greater Equal (x ≥ a), and Greater Than (x > a), where a and/or b are values to be specified. To specify Between or Except Between, two values should be separated by a comma (e.g. "80,90").
- String field
Available conditions are Regex (regular expression) and Equal (exact match).
Once filtered, a '*' mark is displayed on the left side of the annotation track. Additional calls will add filters to the existing one. To reset the filter on the specified segment, choose the segment type and press the Reset This Filter button. To reset all filters, press the Reset All Filters button.

Dynamic Search
Search each genome for specific segments or sets of segments dynamically, by calling a CGI script running on the server. Results will be displayed in the additional annotation tracks, or in the outermost pair of tracks (overwriting the existing data) if the maximum number of segment tracks have already been used.
Currently, the following programs are available:
Blast: BLAST similarity search for a nucleotide [blastn(direct) / tblastx(translation)] or a protein [tblastn] query sequence.
PatSearch: Regular expression pattern search. See the document of the Java regex library.
DirRep: Direct repeat search.
SimpleRep: Simple repeat (or short tandem repeat) search.
View menu
Gene/Segment Data Table
Show table of genes or specified feature segment on the specified genome. In the table, each row contains species name, the beginning position, the ending position, strand direction, assigned color, and the name of the segment. Additional fields are added, if any, depending on the segment type. By clicking a row of this table, the current scope is reset such that the specified gene or segment is centered.
